1. Data preprocessing and curation
This notebook focuses on the preparation of LPS datasets, including both public and private sources, and demonstrates how to merge them for training the PerturbGen model.
1.1. Public LPS data
import gdown # for downloading files from Google Drive
import requests
Downloading the public LPS dataset
url = 'https://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/026/E-MTAB-10026/Files/covid_portal_210320_with_raw.h5ad'
output= './Raw_h5ad'
gdown.download(url,output) # download the file
Downloading...
From: https://ftp.ebi.ac.uk/biostudies/fire/E-MTAB-/026/E-MTAB-10026/Files/covid_portal_210320_with_raw.h5ad
To: /home/jovyan/farm_mount/Cytomeister/Evaluation_datasets/LPS/Public_Emily/Raw_h5ad.zip
100%|██████████| 7.19G/7.19G [01:31<00:00, 78.9MB/s]
'./Raw_h5ad.zip'
Import Libraries
import pandas as pd
import scanpy as sc
import numpy as np
import seaborn as sns
# Read the downloaded h5ad file
adata = sc.read('./Raw.h5ad')
Here, we investigate the .h5ad file to identify LPS samples and healthy control cells in the downloaded LPS data.
adata
AnnData object with n_obs × n_vars = 647366 × 24929
obs: 'sample_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'full_clustering', 'initial_clustering', 'Resample', 'Collection_Day', 'Sex', 'Age_interval', 'Swab_result', 'Status', 'Smoker', 'Status_on_day_collection', 'Status_on_day_collection_summary', 'Days_from_onset', 'Site', 'time_after_LPS', 'Worst_Clinical_Status', 'Outcome', 'patient_id'
var: 'feature_types'
uns: 'hvg', 'leiden', 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_pca_harmony', 'X_umap'
layers: 'raw'
adata.obs['time_after_LPS'].value_counts() # check time points available
time_after_LPS
nan 639482
90m 3999
10h 3885
Name: count, dtype: int64
adata.obs['Status'].value_counts() # check conditions available
Status
Covid 527286
Healthy 97039
Non_covid 15157
LPS 7884
Name: count, dtype: int64
lps_sample_ids = adata.obs.loc[adata.obs['Status'] == 'LPS', 'patient_id'].unique() # get patient ids for LPS samples
healthy_sample_ids = adata.obs.loc[adata.obs['Status'] == 'Healthy', 'patient_id'].unique() # get patient ids for Healthy samples
adata = adata[adata.obs['Status'].isin(['LPS','Healthy'])].copy() # keep only LPS and Healthy samples
Next, we apply a set of modifications and add details to make downstream analyses more convenient e.g. converting time obs into categorical obs
adata.obs['time_after_LPS'] = adata.obs['time_after_LPS'].astype(str) # convert to string
adata.obs['time_after_LPS'] = adata.obs['time_after_LPS'].replace('nan', 'normal') # replace nan with normal
adata.obs['time_after_LPS'] = adata.obs['time_after_LPS'].apply(lambda x: x if x == 'normal' else f'{x}_LPS')
adata.obs['time_after_LPS'] = pd.Categorical(
adata.obs['time_after_LPS'],
categories=['normal', '90m_LPS', '10h_LPS'],
ordered=True
)
adata.obs['time_after_LPS'].value_counts()
time_after_LPS
normal 97039
90m_LPS 3999
10h_LPS 3885
Name: count, dtype: int64
adata.obs['donor_id'] = adata.obs['patient_id']
adata.obs['study'] = 'Public_Emily2021'
adata.obs['Age_interval'].value_counts()
Age_interval
(30, 39] 30538
(50, 59] 26344
(20, 29] 20426
(40, 49] 15926
(60, 69] 9970
(70, 79] 1719
Name: count, dtype: int64
mapping = {
'(20, 29]': 'Adult',
'(30, 39]': 'Adult',
'(40, 49]': 'Adult',
'(50, 59]': 'Adult',
'(60, 69]': 'Adult',
'(70, 79]': 'Old'
}
adata.obs['life_stage'] = adata.obs['Age_interval'].map(mapping)
adata.obs['life_stage'] = pd.Categorical(
adata.obs['life_stage'],
categories=['Embryo', 'Fetal', 'Childhood', 'Young Adult', 'Adult', 'Old'],
ordered=True
)
adata.obs['development_stage'] = adata.obs['life_stage']
adata.obs['tissue'] = 'blood'
adata.X = adata.layers['raw'].copy()
adata.X.max()
np.float32(275279.0)
adata.obs['Resample']
covid_index
AAACCTGAGACCACGA-newcastle65 Initial
AAACCTGAGATGTCGG-newcastle65 Initial
AAACCTGAGGCGATAC-newcastle65 Initial
AAACCTGAGTACACCT-newcastle65 Initial
AAACCTGAGTGAATTG-newcastle65 Initial
...
BGCV15_TTTCCTCTCTGATACG-1 Initial
BGCV15_TTTCCTCTCTTTAGGG-1 Initial
BGCV15_TTTGCGCTCACCGTAA-1 Initial
BGCV15_TTTGGTTTCAAGATCC-1 Initial
BGCV15_TTTGTCACAAGCCATT-1 Initial
Name: Resample, Length: 104923, dtype: category
Categories (1, object): ['Initial']
# Subset to Gene Expression features
adata = adata[:, adata.var['feature_types'] == 'Gene Expression']
ref = pd.read_csv("../../../../Ensembl_symbol_Human_(GRCh38.p14)/mart_export.txt")
adata.var
| feature_types | |
|---|---|
| MIR1302-2HG | Gene Expression |
| AL627309.1 | Gene Expression |
| AL627309.3 | Gene Expression |
| AL627309.2 | Gene Expression |
| AL669831.2 | Gene Expression |
| ... | ... |
| AC007325.2 | Gene Expression |
| AL354822.1 | Gene Expression |
| AC233755.2 | Gene Expression |
| AC233755.1 | Gene Expression |
| AC240274.1 | Gene Expression |
24737 rows × 1 columns
# 1. Convert to string and clean
ref.index = ref.index.astype(str).str.strip().str.upper()
ref["HGNC symbol"] = ref["HGNC symbol"].astype(str).str.strip().str.upper()
# Drop NaNs in gene_symbol (after converting to str 'nan', you can use replace)
ref = ref.replace({"HGNC symbol": {"NAN": None}}).dropna(subset=["HGNC symbol"])
# 2. Map gene_symbol -> ensembl_id by first occurrence
ref_rev = (
ref.reset_index()
.drop_duplicates(subset=["HGNC symbol"])
.set_index("HGNC symbol")["Gene stable ID"]
)
# 3. Map gene_symbol from adata.var_names to ensembl_id
adata.var["gene_symbol"] = adata.var_names.str.strip().str.upper()
adata.var["ensembl_id"] = adata.var["gene_symbol"].map(ref_rev)
# 4. Filter genes without mapping
adata = adata[:, adata.var["ensembl_id"].notna()].copy()
# 5. Set var_names to ensembl_id
adata.var_names = adata.var["ensembl_id"]
print(f"adata now has {adata.n_vars} genes with Ensembl IDs as var_names.")
/tmp/ipykernel_962786/4272093940.py:16: ImplicitModificationWarning: Trying to modify attribute `.var` of view, initializing view as actual.
adata.var["gene_symbol"] = adata.var_names.str.strip().str.upper()
adata now has 19193 genes with Ensembl IDs as var_names.
adata.var.index.name = 'gene_id'
adata.var
| feature_types | gene_symbol | ensembl_id | |
|---|---|---|---|
| gene_id | |||
| ENSG00000243485 | Gene Expression | MIR1302-2HG | ENSG00000243485 |
| ENSG00000177757 | Gene Expression | FAM87B | ENSG00000177757 |
| ENSG00000225880 | Gene Expression | LINC00115 | ENSG00000225880 |
| ENSG00000230368 | Gene Expression | FAM41C | ENSG00000230368 |
| ENSG00000187634 | Gene Expression | SAMD11 | ENSG00000187634 |
| ... | ... | ... | ... |
| ENSG00000198886 | Gene Expression | MT-ND4 | ENSG00000198886 |
| ENSG00000198786 | Gene Expression | MT-ND5 | ENSG00000198786 |
| ENSG00000198695 | Gene Expression | MT-ND6 | ENSG00000198695 |
| ENSG00000198727 | Gene Expression | MT-CYB | ENSG00000198727 |
| ENSG00000274847 | Gene Expression | MAFIP | ENSG00000274847 |
19193 rows × 3 columns
adata.obs['development_stage']
covid_index
AAACCTGAGACCACGA-newcastle65 Adult
AAACCTGAGATGTCGG-newcastle65 Adult
AAACCTGAGGCGATAC-newcastle65 Adult
AAACCTGAGTACACCT-newcastle65 Adult
AAACCTGAGTGAATTG-newcastle65 Adult
...
BGCV15_TTTCCTCTCTGATACG-1 Adult
BGCV15_TTTCCTCTCTTTAGGG-1 Adult
BGCV15_TTTGCGCTCACCGTAA-1 Adult
BGCV15_TTTGGTTTCAAGATCC-1 Adult
BGCV15_TTTGTCACAAGCCATT-1 Adult
Name: development_stage, Length: 104923, dtype: category
Categories (2, object): ['Adult' < 'Old']
adata.obs['full_clustering'].value_counts()
full_clustering
CD4.Naive 13829
NK_16hi 13473
CD8.Naive 8105
CD4.CM 7505
B_naive 6908
CD83_CD14_mono 6772
CD8.TE 6501
CD4.IL22 6379
CD8.EM 5734
gdT 4948
CD14_mono 3774
MAIT 3465
CD16_mono 3007
Platelets 2363
NK_56hi 2190
B_switched_memory 1395
DC2 1052
DC3 975
B_immature 966
NKT 864
CD4.Tfh 800
pDC 678
B_non-switched_memory 647
B_exhausted 334
RBC 301
NK_prolif 221
ILC1_3 213
C1_CD16_mono 198
CD4.EM 188
Plasma_cell_IgA 173
Plasma_cell_IgG 168
DC1 104
CD4.Th1 101
Plasmablast 95
Plasma_cell_IgM 84
Treg 75
HSC_erythroid 74
CD8.Prolif 70
HSC_CD38pos 61
ILC2 36
CD4.Prolif 32
CD4.Th2 16
ASDC 16
HSC_CD38neg 13
DC_prolif 6
HSC_prolif 6
Mono_prolif 4
HSC_myeloid 3
CD4.Th17 1
Name: count, dtype: int64
adata.obs['cell_type'] = adata.obs['full_clustering'].copy()
cell_type_counts = adata.obs['cell_type'].value_counts()
valid_cell_types = cell_type_counts[cell_type_counts >= 50].index
adata = adata[adata.obs['cell_type'].isin(valid_cell_types)].copy()
adata.obs['time_after_LPS']
covid_index
AAACCTGAGACCACGA-newcastle65 normal
AAACCTGAGATGTCGG-newcastle65 normal
AAACCTGAGGCGATAC-newcastle65 normal
AAACCTGAGTACACCT-newcastle65 normal
AAACCTGAGTGAATTG-newcastle65 normal
...
BGCV15_TTTCCTCTCTGATACG-1 normal
BGCV15_TTTCCTCTCTTTAGGG-1 normal
BGCV15_TTTGCGCTCACCGTAA-1 normal
BGCV15_TTTGGTTTCAAGATCC-1 normal
BGCV15_TTTGTCACAAGCCATT-1 normal
Name: time_after_LPS, Length: 104790, dtype: category
Categories (3, object): ['normal' < '90m_LPS' < '10h_LPS']
# If data is sparse, it's best to use .X.sum(axis=1).A1
if hasattr(adata.X, 'sum'):
adata.obs['n_counts'] = np.ravel(adata.X.sum(axis=1))
else:
adata.obs['n_counts'] = adata.X.sum(axis=1)
adata.obs['n_counts']
covid_index
AAACCTGAGACCACGA-newcastle65 4242.0
AAACCTGAGATGTCGG-newcastle65 4708.0
AAACCTGAGGCGATAC-newcastle65 2858.0
AAACCTGAGTACACCT-newcastle65 5227.0
AAACCTGAGTGAATTG-newcastle65 4012.0
...
BGCV15_TTTCCTCTCTGATACG-1 5766.0
BGCV15_TTTCCTCTCTTTAGGG-1 7415.0
BGCV15_TTTGCGCTCACCGTAA-1 5168.0
BGCV15_TTTGGTTTCAAGATCC-1 6504.0
BGCV15_TTTGTCACAAGCCATT-1 5246.0
Name: n_counts, Length: 104790, dtype: float32
Eventually, we save the public data
adata.write_h5ad('./Public_LPS_PP.h5ad')
1.2. Private LPS data
This data will be published after the paper publication.
tcell=sc.read_h5ad('/path/to/data1/tcell_raw_matrix_annotated.h5ad')
tcell
AnnData object with n_obs × n_vars = 69370 × 35976
obs: 'patient_id', 'time_after_LPS', 'Sex', 'age', 'batch', 'IR_VJ_1_locus_tcr', 'IR_VJ_2_locus_tcr', 'IR_VDJ_1_locus_tcr', 'IR_VDJ_2_locus_tcr', 'IR_VJ_1_cdr3_tcr', 'IR_VJ_2_cdr3_tcr', 'IR_VDJ_1_cdr3_tcr', 'IR_VDJ_2_cdr3_tcr', 'IR_VJ_1_cdr3_nt_tcr', 'IR_VJ_2_cdr3_nt_tcr', 'IR_VDJ_1_cdr3_nt_tcr', 'IR_VDJ_2_cdr3_nt_tcr', 'IR_VJ_1_expr_tcr', 'IR_VJ_2_expr_tcr', 'IR_VDJ_1_expr_tcr', 'IR_VDJ_2_expr_tcr', 'IR_VJ_1_expr_raw_tcr', 'IR_VJ_2_expr_raw_tcr', 'IR_VDJ_1_expr_raw_tcr', 'IR_VDJ_2_expr_raw_tcr', 'IR_VJ_1_v_gene_tcr', 'IR_VJ_2_v_gene_tcr', 'IR_VDJ_1_v_gene_tcr', 'IR_VDJ_2_v_gene_tcr', 'IR_VJ_1_d_gene_tcr', 'IR_VJ_2_d_gene_tcr', 'IR_VDJ_1_d_gene_tcr', 'IR_VDJ_2_d_gene_tcr', 'IR_VJ_1_j_gene_tcr', 'IR_VJ_2_j_gene_tcr', 'IR_VDJ_1_j_gene_tcr', 'IR_VDJ_2_j_gene_tcr', 'IR_VJ_1_c_gene_tcr', 'IR_VJ_2_c_gene_tcr', 'IR_VDJ_1_c_gene_tcr', 'IR_VDJ_2_c_gene_tcr', 'IR_VJ_1_junction_ins_tcr', 'IR_VJ_2_junction_ins_tcr', 'IR_VDJ_1_junction_ins_tcr', 'IR_VDJ_2_junction_ins_tcr', 'has_ir_tcr', 'multi_chain_tcr', 'IR_VJ_1_locus_bcr', 'IR_VJ_2_locus_bcr', 'IR_VDJ_1_locus_bcr', 'IR_VDJ_2_locus_bcr', 'IR_VJ_1_cdr3_bcr', 'IR_VJ_2_cdr3_bcr', 'IR_VDJ_1_cdr3_bcr', 'IR_VDJ_2_cdr3_bcr', 'IR_VJ_1_cdr3_nt_bcr', 'IR_VJ_2_cdr3_nt_bcr', 'IR_VDJ_1_cdr3_nt_bcr', 'IR_VDJ_2_cdr3_nt_bcr', 'IR_VJ_1_expr_bcr', 'IR_VJ_2_expr_bcr', 'IR_VDJ_1_expr_bcr', 'IR_VDJ_2_expr_bcr', 'IR_VJ_1_expr_raw_bcr', 'IR_VJ_2_expr_raw_bcr', 'IR_VDJ_1_expr_raw_bcr', 'IR_VDJ_2_expr_raw_bcr', 'IR_VJ_1_v_gene_bcr', 'IR_VJ_2_v_gene_bcr', 'IR_VDJ_1_v_gene_bcr', 'IR_VDJ_2_v_gene_bcr', 'IR_VJ_1_d_gene_bcr', 'IR_VJ_2_d_gene_bcr', 'IR_VDJ_1_d_gene_bcr', 'IR_VDJ_2_d_gene_bcr', 'IR_VJ_1_j_gene_bcr', 'IR_VJ_2_j_gene_bcr', 'IR_VDJ_1_j_gene_bcr', 'IR_VDJ_2_j_gene_bcr', 'IR_VJ_1_c_gene_bcr', 'IR_VJ_2_c_gene_bcr', 'IR_VDJ_1_c_gene_bcr', 'IR_VDJ_2_c_gene_bcr', 'IR_VJ_1_junction_ins_bcr', 'IR_VJ_2_junction_ins_bcr', 'IR_VDJ_1_junction_ins_bcr', 'IR_VDJ_2_junction_ins_bcr', 'has_ir_bcr', 'multi_chain_bcr', 'scr_th', 'doublet_scores', 'cal_th', 'scr_predicted_doublets', 'cal_dobulets', 'data_batch', 'individual_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'cell_typist_full_predicted_labels', 'cell_typist_full_majority_voting', 'cell_typist_initial_predicted_labels', 'cell_typist_initial_majority_voting', 'final_anno', 'timeline', 'sample_id', 'all_T'
var: 'gene_ids', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'GEX'
bcell=sc.read_h5ad('/path/to/data2/bcell_raw_matrix_annotated_allanno.h5ad')
bcell
AnnData object with n_obs × n_vars = 11964 × 36601
obs: 'patient_id', 'time_after_LPS', 'Sex', 'age', 'batch', 'IR_VJ_1_locus_tcr', 'IR_VJ_2_locus_tcr', 'IR_VDJ_1_locus_tcr', 'IR_VDJ_2_locus_tcr', 'IR_VJ_1_cdr3_tcr', 'IR_VJ_2_cdr3_tcr', 'IR_VDJ_1_cdr3_tcr', 'IR_VDJ_2_cdr3_tcr', 'IR_VJ_1_cdr3_nt_tcr', 'IR_VJ_2_cdr3_nt_tcr', 'IR_VDJ_1_cdr3_nt_tcr', 'IR_VDJ_2_cdr3_nt_tcr', 'IR_VJ_1_expr_tcr', 'IR_VJ_2_expr_tcr', 'IR_VDJ_1_expr_tcr', 'IR_VDJ_2_expr_tcr', 'IR_VJ_1_expr_raw_tcr', 'IR_VJ_2_expr_raw_tcr', 'IR_VDJ_1_expr_raw_tcr', 'IR_VDJ_2_expr_raw_tcr', 'IR_VJ_1_v_gene_tcr', 'IR_VJ_2_v_gene_tcr', 'IR_VDJ_1_v_gene_tcr', 'IR_VDJ_2_v_gene_tcr', 'IR_VJ_1_d_gene_tcr', 'IR_VJ_2_d_gene_tcr', 'IR_VDJ_1_d_gene_tcr', 'IR_VDJ_2_d_gene_tcr', 'IR_VJ_1_j_gene_tcr', 'IR_VJ_2_j_gene_tcr', 'IR_VDJ_1_j_gene_tcr', 'IR_VDJ_2_j_gene_tcr', 'IR_VJ_1_c_gene_tcr', 'IR_VJ_2_c_gene_tcr', 'IR_VDJ_1_c_gene_tcr', 'IR_VDJ_2_c_gene_tcr', 'IR_VJ_1_junction_ins_tcr', 'IR_VJ_2_junction_ins_tcr', 'IR_VDJ_1_junction_ins_tcr', 'IR_VDJ_2_junction_ins_tcr', 'has_ir_tcr', 'multi_chain_tcr', 'IR_VJ_1_locus_bcr', 'IR_VJ_2_locus_bcr', 'IR_VDJ_1_locus_bcr', 'IR_VDJ_2_locus_bcr', 'IR_VJ_1_cdr3_bcr', 'IR_VJ_2_cdr3_bcr', 'IR_VDJ_1_cdr3_bcr', 'IR_VDJ_2_cdr3_bcr', 'IR_VJ_1_cdr3_nt_bcr', 'IR_VJ_2_cdr3_nt_bcr', 'IR_VDJ_1_cdr3_nt_bcr', 'IR_VDJ_2_cdr3_nt_bcr', 'IR_VJ_1_expr_bcr', 'IR_VJ_2_expr_bcr', 'IR_VDJ_1_expr_bcr', 'IR_VDJ_2_expr_bcr', 'IR_VJ_1_expr_raw_bcr', 'IR_VJ_2_expr_raw_bcr', 'IR_VDJ_1_expr_raw_bcr', 'IR_VDJ_2_expr_raw_bcr', 'IR_VJ_1_v_gene_bcr', 'IR_VJ_2_v_gene_bcr', 'IR_VDJ_1_v_gene_bcr', 'IR_VDJ_2_v_gene_bcr', 'IR_VJ_1_d_gene_bcr', 'IR_VJ_2_d_gene_bcr', 'IR_VDJ_1_d_gene_bcr', 'IR_VDJ_2_d_gene_bcr', 'IR_VJ_1_j_gene_bcr', 'IR_VJ_2_j_gene_bcr', 'IR_VDJ_1_j_gene_bcr', 'IR_VDJ_2_j_gene_bcr', 'IR_VJ_1_c_gene_bcr', 'IR_VJ_2_c_gene_bcr', 'IR_VDJ_1_c_gene_bcr', 'IR_VDJ_2_c_gene_bcr', 'IR_VJ_1_junction_ins_bcr', 'IR_VJ_2_junction_ins_bcr', 'IR_VDJ_1_junction_ins_bcr', 'IR_VDJ_2_junction_ins_bcr', 'has_ir_bcr', 'multi_chain_bcr', 'scr_th', 'doublet_scores', 'cal_th', 'scr_predicted_doublets', 'cal_dobulets', 'data_batch', 'individual_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'cell_typist_full_predicted_labels', 'cell_typist_full_majority_voting', 'cell_typist_initial_predicted_labels', 'cell_typist_initial_majority_voting', 'final_anno', 'final_anno1', 'timeline', 'sample_id', 'all_B'
var: 'gene_ids', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
myeloid=sc.read_h5ad('/path/to/data3/mcell_raw_matrix_annotated.h5ad')
combined_data=tcell.concatenate(bcell,myeloid) # combine the three datasets
/tmp/ipykernel_968620/3716584077.py:1: FutureWarning: Use anndata.concat instead of AnnData.concatenate, AnnData.concatenate is deprecated and will be removed in the future. See the tutorial for concat at: https://anndata.readthedocs.io/en/latest/concatenation.html
combined_data=tcell.concatenate(bcell,myeloid)
combined_data # check combined data
AnnData object with n_obs × n_vars = 122263 × 35976
obs: 'patient_id', 'time_after_LPS', 'Sex', 'age', 'batch', 'IR_VJ_1_locus_tcr', 'IR_VJ_2_locus_tcr', 'IR_VDJ_1_locus_tcr', 'IR_VDJ_2_locus_tcr', 'IR_VJ_1_cdr3_tcr', 'IR_VJ_2_cdr3_tcr', 'IR_VDJ_1_cdr3_tcr', 'IR_VDJ_2_cdr3_tcr', 'IR_VJ_1_cdr3_nt_tcr', 'IR_VJ_2_cdr3_nt_tcr', 'IR_VDJ_1_cdr3_nt_tcr', 'IR_VDJ_2_cdr3_nt_tcr', 'IR_VJ_1_expr_tcr', 'IR_VJ_2_expr_tcr', 'IR_VDJ_1_expr_tcr', 'IR_VDJ_2_expr_tcr', 'IR_VJ_1_expr_raw_tcr', 'IR_VJ_2_expr_raw_tcr', 'IR_VDJ_1_expr_raw_tcr', 'IR_VDJ_2_expr_raw_tcr', 'IR_VJ_1_v_gene_tcr', 'IR_VJ_2_v_gene_tcr', 'IR_VDJ_1_v_gene_tcr', 'IR_VDJ_2_v_gene_tcr', 'IR_VJ_1_d_gene_tcr', 'IR_VJ_2_d_gene_tcr', 'IR_VDJ_1_d_gene_tcr', 'IR_VDJ_2_d_gene_tcr', 'IR_VJ_1_j_gene_tcr', 'IR_VJ_2_j_gene_tcr', 'IR_VDJ_1_j_gene_tcr', 'IR_VDJ_2_j_gene_tcr', 'IR_VJ_1_c_gene_tcr', 'IR_VJ_2_c_gene_tcr', 'IR_VDJ_1_c_gene_tcr', 'IR_VDJ_2_c_gene_tcr', 'IR_VJ_1_junction_ins_tcr', 'IR_VJ_2_junction_ins_tcr', 'IR_VDJ_1_junction_ins_tcr', 'IR_VDJ_2_junction_ins_tcr', 'has_ir_tcr', 'multi_chain_tcr', 'IR_VJ_1_locus_bcr', 'IR_VJ_2_locus_bcr', 'IR_VDJ_1_locus_bcr', 'IR_VDJ_2_locus_bcr', 'IR_VJ_1_cdr3_bcr', 'IR_VJ_2_cdr3_bcr', 'IR_VDJ_1_cdr3_bcr', 'IR_VDJ_2_cdr3_bcr', 'IR_VJ_1_cdr3_nt_bcr', 'IR_VJ_2_cdr3_nt_bcr', 'IR_VDJ_1_cdr3_nt_bcr', 'IR_VDJ_2_cdr3_nt_bcr', 'IR_VJ_1_expr_bcr', 'IR_VJ_2_expr_bcr', 'IR_VDJ_1_expr_bcr', 'IR_VDJ_2_expr_bcr', 'IR_VJ_1_expr_raw_bcr', 'IR_VJ_2_expr_raw_bcr', 'IR_VDJ_1_expr_raw_bcr', 'IR_VDJ_2_expr_raw_bcr', 'IR_VJ_1_v_gene_bcr', 'IR_VJ_2_v_gene_bcr', 'IR_VDJ_1_v_gene_bcr', 'IR_VDJ_2_v_gene_bcr', 'IR_VJ_1_d_gene_bcr', 'IR_VJ_2_d_gene_bcr', 'IR_VDJ_1_d_gene_bcr', 'IR_VDJ_2_d_gene_bcr', 'IR_VJ_1_j_gene_bcr', 'IR_VJ_2_j_gene_bcr', 'IR_VDJ_1_j_gene_bcr', 'IR_VDJ_2_j_gene_bcr', 'IR_VJ_1_c_gene_bcr', 'IR_VJ_2_c_gene_bcr', 'IR_VDJ_1_c_gene_bcr', 'IR_VDJ_2_c_gene_bcr', 'IR_VJ_1_junction_ins_bcr', 'IR_VJ_2_junction_ins_bcr', 'IR_VDJ_1_junction_ins_bcr', 'IR_VDJ_2_junction_ins_bcr', 'has_ir_bcr', 'multi_chain_bcr', 'scr_th', 'doublet_scores', 'cal_th', 'scr_predicted_doublets', 'cal_dobulets', 'data_batch', 'individual_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'cell_typist_full_predicted_labels', 'cell_typist_full_majority_voting', 'cell_typist_initial_predicted_labels', 'cell_typist_initial_majority_voting', 'final_anno', 'timeline', 'sample_id', 'all_T', 'final_anno1', 'all_B'
var: 'gene_ids', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'GEX-0', 'GEX-2'
Next, we apply a set of modifications, QC analyses and preprocessing as data is raw
# mitochondrial genes
combined_data.var["mt"] = combined_data.var_names.str.startswith("MT-")
# ribosomal genes
combined_data.var["ribo"] = combined_data.var_names.str.startswith(("RPS", "RPL"))
# hemoglobin genes.
combined_data.var["hb"] = combined_data.var_names.str.contains(("^HB[^(P)]"))
sc.pp.calculate_qc_metrics( # calculate QC metrics
combined_data, qc_vars=["mt", "ribo", "hb"], inplace=True, percent_top=[20], log1p=True
)
combined_data
AnnData object with n_obs × n_vars = 122263 × 35976
obs: 'patient_id', 'time_after_LPS', 'Sex', 'age', 'batch', 'IR_VJ_1_locus_tcr', 'IR_VJ_2_locus_tcr', 'IR_VDJ_1_locus_tcr', 'IR_VDJ_2_locus_tcr', 'IR_VJ_1_cdr3_tcr', 'IR_VJ_2_cdr3_tcr', 'IR_VDJ_1_cdr3_tcr', 'IR_VDJ_2_cdr3_tcr', 'IR_VJ_1_cdr3_nt_tcr', 'IR_VJ_2_cdr3_nt_tcr', 'IR_VDJ_1_cdr3_nt_tcr', 'IR_VDJ_2_cdr3_nt_tcr', 'IR_VJ_1_expr_tcr', 'IR_VJ_2_expr_tcr', 'IR_VDJ_1_expr_tcr', 'IR_VDJ_2_expr_tcr', 'IR_VJ_1_expr_raw_tcr', 'IR_VJ_2_expr_raw_tcr', 'IR_VDJ_1_expr_raw_tcr', 'IR_VDJ_2_expr_raw_tcr', 'IR_VJ_1_v_gene_tcr', 'IR_VJ_2_v_gene_tcr', 'IR_VDJ_1_v_gene_tcr', 'IR_VDJ_2_v_gene_tcr', 'IR_VJ_1_d_gene_tcr', 'IR_VJ_2_d_gene_tcr', 'IR_VDJ_1_d_gene_tcr', 'IR_VDJ_2_d_gene_tcr', 'IR_VJ_1_j_gene_tcr', 'IR_VJ_2_j_gene_tcr', 'IR_VDJ_1_j_gene_tcr', 'IR_VDJ_2_j_gene_tcr', 'IR_VJ_1_c_gene_tcr', 'IR_VJ_2_c_gene_tcr', 'IR_VDJ_1_c_gene_tcr', 'IR_VDJ_2_c_gene_tcr', 'IR_VJ_1_junction_ins_tcr', 'IR_VJ_2_junction_ins_tcr', 'IR_VDJ_1_junction_ins_tcr', 'IR_VDJ_2_junction_ins_tcr', 'has_ir_tcr', 'multi_chain_tcr', 'IR_VJ_1_locus_bcr', 'IR_VJ_2_locus_bcr', 'IR_VDJ_1_locus_bcr', 'IR_VDJ_2_locus_bcr', 'IR_VJ_1_cdr3_bcr', 'IR_VJ_2_cdr3_bcr', 'IR_VDJ_1_cdr3_bcr', 'IR_VDJ_2_cdr3_bcr', 'IR_VJ_1_cdr3_nt_bcr', 'IR_VJ_2_cdr3_nt_bcr', 'IR_VDJ_1_cdr3_nt_bcr', 'IR_VDJ_2_cdr3_nt_bcr', 'IR_VJ_1_expr_bcr', 'IR_VJ_2_expr_bcr', 'IR_VDJ_1_expr_bcr', 'IR_VDJ_2_expr_bcr', 'IR_VJ_1_expr_raw_bcr', 'IR_VJ_2_expr_raw_bcr', 'IR_VDJ_1_expr_raw_bcr', 'IR_VDJ_2_expr_raw_bcr', 'IR_VJ_1_v_gene_bcr', 'IR_VJ_2_v_gene_bcr', 'IR_VDJ_1_v_gene_bcr', 'IR_VDJ_2_v_gene_bcr', 'IR_VJ_1_d_gene_bcr', 'IR_VJ_2_d_gene_bcr', 'IR_VDJ_1_d_gene_bcr', 'IR_VDJ_2_d_gene_bcr', 'IR_VJ_1_j_gene_bcr', 'IR_VJ_2_j_gene_bcr', 'IR_VDJ_1_j_gene_bcr', 'IR_VDJ_2_j_gene_bcr', 'IR_VJ_1_c_gene_bcr', 'IR_VJ_2_c_gene_bcr', 'IR_VDJ_1_c_gene_bcr', 'IR_VDJ_2_c_gene_bcr', 'IR_VJ_1_junction_ins_bcr', 'IR_VJ_2_junction_ins_bcr', 'IR_VDJ_1_junction_ins_bcr', 'IR_VDJ_2_junction_ins_bcr', 'has_ir_bcr', 'multi_chain_bcr', 'scr_th', 'doublet_scores', 'cal_th', 'scr_predicted_doublets', 'cal_dobulets', 'data_batch', 'individual_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'cell_typist_full_predicted_labels', 'cell_typist_full_majority_voting', 'cell_typist_initial_predicted_labels', 'cell_typist_initial_majority_voting', 'final_anno', 'timeline', 'sample_id', 'all_T', 'final_anno1', 'all_B', 'log1p_n_genes_by_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'log1p_total_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb'
var: 'gene_ids', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'GEX-0', 'GEX-2', 'ribo', 'hb', 'log1p_mean_counts', 'log1p_total_counts'
Visualize QC metrics
p1 = sns.displot(combined_data.obs["total_counts"], bins=100, kde=False)
sc.pl.violin(combined_data, 'total_counts')
p2 = sc.pl.violin(combined_data, "pct_counts_mt")
p3 = sc.pl.scatter(combined_data, "total_counts", "n_genes_by_counts", color="pct_counts_mt")
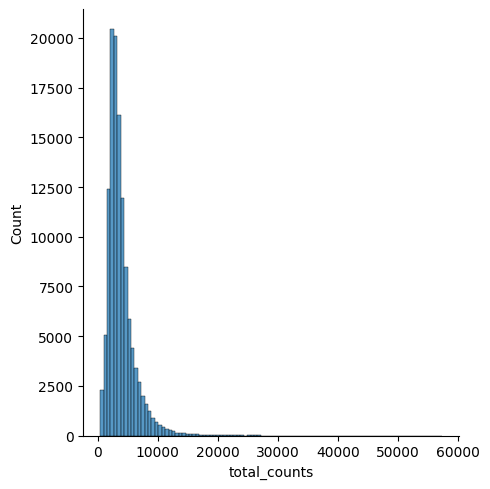
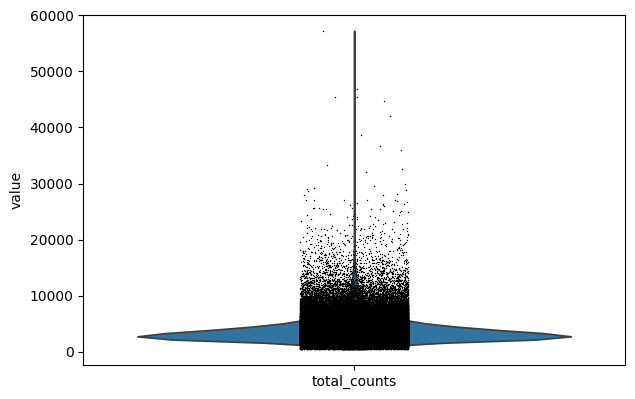
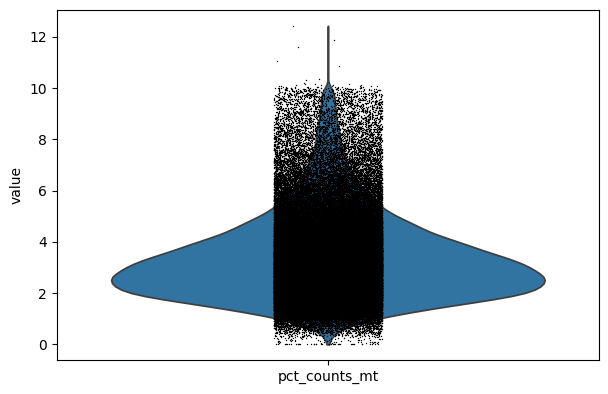
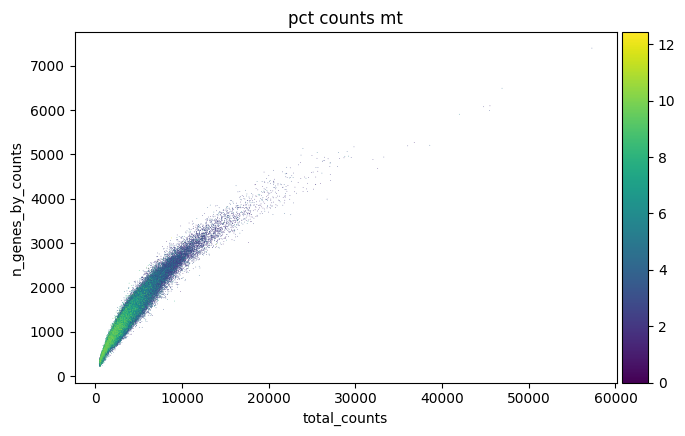
combined_data = combined_data[combined_data.obs['pct_counts_mt'] <= 8] # filter cells with high mitochondrial content
sc.pp.filter_cells(combined_data, max_counts=150000) # filter cells with too many counts
/software/cellgen/team361/am74/envs/pertpy/lib/python3.9/site-packages/scanpy/preprocessing/_simple.py:165: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
adata.obs["n_counts"] = number
combined_data.obs['n_genes'] = (combined_data.X > 0).sum(1) # calculate number of genes per cell
sns.distplot(combined_data.obs['n_genes'], kde=False, bins=60) # visualize number of genes per cell
/tmp/ipykernel_968620/3987137127.py:2: UserWarning:
`distplot` is a deprecated function and will be removed in seaborn v0.14.0.
Please adapt your code to use either `displot` (a figure-level function with
similar flexibility) or `histplot` (an axes-level function for histograms).
For a guide to updating your code to use the new functions, please see
https://gist.github.com/mwaskom/de44147ed2974457ad6372750bbe5751
sns.distplot(combined_data.obs['n_genes'], kde=False, bins=60)
<Axes: xlabel='n_genes'>
sc.pp.filter_genes(combined_data, min_cells=20) # filter genes expressed in less than 20 cells
print('Number of genes after cell filter: {:d}'.format(combined_data.n_vars))
Number of genes after cell filter: 19186
combined_data.obs['batch'] = combined_data.obs['batch'].cat.rename_categories({
'0': 'T cell lineage',
'1': 'B cell lineage',
'2': 'Myeloid lineage'
})
combined_data.obs['final_anno'].value_counts()
final_anno
CD14mono 23888
CD4_naive 18268
CD8_naive 13249
CD4_memory 10259
CD8_EM 8869
B_naive 8185
Int.mono 7229
NK_CD56dim 4922
CD14mono_M1 4129
gdT 3325
MAIT 2577
Treg 2382
NKT 2376
non-swtiched memory 1583
CD16mono 1213
switched memory 1158
CD8_CM 1004
DC2 972
Platelet 862
NK_CD56bright 856
pDC 696
DC3 488
non-switched memory/CD11c 301
HSPC 227
plasma_IgA 160
DC1 60
plasma_dividing 50
plasma_IgG 31
ASDC 23
plasma_IgM 18
Name: count, dtype: int64
combined_data.X.max() # check max counts after filtering
2717.0
combined_data.obs['cell_type'] = combined_data.obs['final_anno'] # set cell_type based on final_anno
combined_data.obs['patient_id']
AAACCTGAGACAAAGG-1-0 IVLPS01_Baseline
AAACCTGAGCGTTCCG-1-0 IVLPS01_Baseline
AAACCTGAGTGTTTGC-1-0 IVLPS01_Baseline
AAACCTGCAAACTGTC-1-0 IVLPS01_Baseline
AAACCTGCACAACGTT-1-0 IVLPS01_Baseline
...
TTTGGTTTCGGAATCT-1-2 IVLPS12_10h
TTTGTCAAGGCATGTG-1-2 IVLPS12_10h
TTTGTCACAAAGGAAG-1-2 IVLPS12_10h
TTTGTCATCAACACTG-1-2 IVLPS12_10h
TTTGTCATCACAACGT-1-2 IVLPS12_10h
Name: patient_id, Length: 119360, dtype: category
Categories (23, object): ['IVLPS01_10h', 'IVLPS01_6h', 'IVLPS01_90min', 'IVLPS01_Baseline', ..., 'IVLPS06_Baseline', 'IVLPS12_10h', 'IVLPS12_6h', 'IVLPS12_90min']
combined_data.obs['donor_id'] = combined_data.obs['patient_id'].copy()
combined_data.obs['time_after_LPS']
AAACCTGAGACAAAGG-1-0 0m
AAACCTGAGCGTTCCG-1-0 0m
AAACCTGAGTGTTTGC-1-0 0m
AAACCTGCAAACTGTC-1-0 0m
AAACCTGCACAACGTT-1-0 0m
...
TTTGGTTTCGGAATCT-1-2 10h
TTTGTCAAGGCATGTG-1-2 10h
TTTGTCACAAAGGAAG-1-2 10h
TTTGTCATCAACACTG-1-2 10h
TTTGTCATCACAACGT-1-2 10h
Name: time_after_LPS, Length: 119360, dtype: category
Categories (4, object): ['0m', '10h', '6h', '90m']
combined_data.obs['time_after_LPS'] = combined_data.obs['time_after_LPS'].cat.rename_categories({
'0m': 'normal',
'10h': '10h_LPS',
'6h': '6h_LPS',
'90m': '90m_LPS'
})
combined_data.obs['time_after_LPS']
AAACCTGAGACAAAGG-1-0 normal
AAACCTGAGCGTTCCG-1-0 normal
AAACCTGAGTGTTTGC-1-0 normal
AAACCTGCAAACTGTC-1-0 normal
AAACCTGCACAACGTT-1-0 normal
...
TTTGGTTTCGGAATCT-1-2 10h_LPS
TTTGTCAAGGCATGTG-1-2 10h_LPS
TTTGTCACAAAGGAAG-1-2 10h_LPS
TTTGTCATCAACACTG-1-2 10h_LPS
TTTGTCATCACAACGT-1-2 10h_LPS
Name: time_after_LPS, Length: 119360, dtype: category
Categories (4, object): ['normal', '10h_LPS', '6h_LPS', '90m_LPS']
combined_data.obs['study'] = 'Private_Data_Emily'
combined_data.obs['tissue'] = 'blood'
combined_data
AnnData object with n_obs × n_vars = 119360 × 19186
obs: 'patient_id', 'time_after_LPS', 'Sex', 'age', 'batch', 'IR_VJ_1_locus_tcr', 'IR_VJ_2_locus_tcr', 'IR_VDJ_1_locus_tcr', 'IR_VDJ_2_locus_tcr', 'IR_VJ_1_cdr3_tcr', 'IR_VJ_2_cdr3_tcr', 'IR_VDJ_1_cdr3_tcr', 'IR_VDJ_2_cdr3_tcr', 'IR_VJ_1_cdr3_nt_tcr', 'IR_VJ_2_cdr3_nt_tcr', 'IR_VDJ_1_cdr3_nt_tcr', 'IR_VDJ_2_cdr3_nt_tcr', 'IR_VJ_1_expr_tcr', 'IR_VJ_2_expr_tcr', 'IR_VDJ_1_expr_tcr', 'IR_VDJ_2_expr_tcr', 'IR_VJ_1_expr_raw_tcr', 'IR_VJ_2_expr_raw_tcr', 'IR_VDJ_1_expr_raw_tcr', 'IR_VDJ_2_expr_raw_tcr', 'IR_VJ_1_v_gene_tcr', 'IR_VJ_2_v_gene_tcr', 'IR_VDJ_1_v_gene_tcr', 'IR_VDJ_2_v_gene_tcr', 'IR_VJ_1_d_gene_tcr', 'IR_VJ_2_d_gene_tcr', 'IR_VDJ_1_d_gene_tcr', 'IR_VDJ_2_d_gene_tcr', 'IR_VJ_1_j_gene_tcr', 'IR_VJ_2_j_gene_tcr', 'IR_VDJ_1_j_gene_tcr', 'IR_VDJ_2_j_gene_tcr', 'IR_VJ_1_c_gene_tcr', 'IR_VJ_2_c_gene_tcr', 'IR_VDJ_1_c_gene_tcr', 'IR_VDJ_2_c_gene_tcr', 'IR_VJ_1_junction_ins_tcr', 'IR_VJ_2_junction_ins_tcr', 'IR_VDJ_1_junction_ins_tcr', 'IR_VDJ_2_junction_ins_tcr', 'has_ir_tcr', 'multi_chain_tcr', 'IR_VJ_1_locus_bcr', 'IR_VJ_2_locus_bcr', 'IR_VDJ_1_locus_bcr', 'IR_VDJ_2_locus_bcr', 'IR_VJ_1_cdr3_bcr', 'IR_VJ_2_cdr3_bcr', 'IR_VDJ_1_cdr3_bcr', 'IR_VDJ_2_cdr3_bcr', 'IR_VJ_1_cdr3_nt_bcr', 'IR_VJ_2_cdr3_nt_bcr', 'IR_VDJ_1_cdr3_nt_bcr', 'IR_VDJ_2_cdr3_nt_bcr', 'IR_VJ_1_expr_bcr', 'IR_VJ_2_expr_bcr', 'IR_VDJ_1_expr_bcr', 'IR_VDJ_2_expr_bcr', 'IR_VJ_1_expr_raw_bcr', 'IR_VJ_2_expr_raw_bcr', 'IR_VDJ_1_expr_raw_bcr', 'IR_VDJ_2_expr_raw_bcr', 'IR_VJ_1_v_gene_bcr', 'IR_VJ_2_v_gene_bcr', 'IR_VDJ_1_v_gene_bcr', 'IR_VDJ_2_v_gene_bcr', 'IR_VJ_1_d_gene_bcr', 'IR_VJ_2_d_gene_bcr', 'IR_VDJ_1_d_gene_bcr', 'IR_VDJ_2_d_gene_bcr', 'IR_VJ_1_j_gene_bcr', 'IR_VJ_2_j_gene_bcr', 'IR_VDJ_1_j_gene_bcr', 'IR_VDJ_2_j_gene_bcr', 'IR_VJ_1_c_gene_bcr', 'IR_VJ_2_c_gene_bcr', 'IR_VDJ_1_c_gene_bcr', 'IR_VDJ_2_c_gene_bcr', 'IR_VJ_1_junction_ins_bcr', 'IR_VJ_2_junction_ins_bcr', 'IR_VDJ_1_junction_ins_bcr', 'IR_VDJ_2_junction_ins_bcr', 'has_ir_bcr', 'multi_chain_bcr', 'scr_th', 'doublet_scores', 'cal_th', 'scr_predicted_doublets', 'cal_dobulets', 'data_batch', 'individual_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'cell_typist_full_predicted_labels', 'cell_typist_full_majority_voting', 'cell_typist_initial_predicted_labels', 'cell_typist_initial_majority_voting', 'final_anno', 'timeline', 'sample_id', 'all_T', 'final_anno1', 'all_B', 'log1p_n_genes_by_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'log1p_total_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'n_counts', 'cell_type', 'donor_id', 'study', 'tissue'
var: 'gene_ids', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'GEX-0', 'GEX-2', 'ribo', 'hb', 'log1p_mean_counts', 'log1p_total_counts', 'n_cells'
combined_data.obs['age'].value_counts()
age
23.0 29443
37.0 29386
32.0 21621
36.0 18532
29.0 15735
24.0 4643
Name: count, dtype: int64
def age_to_group(age):
if pd.isna(age):
return 'Unknown'
elif age < 0:
# Prenatal age, optional handling
return 'Prenatal'
elif age <= 18:
return 'Childhood'
elif 18 < age <= 25:
return 'Young Adult'
elif 26 <= age <= 64:
return 'Adult'
elif age >= 65:
return 'Old'
else:
return 'Unknown'
combined_data.obs['development_stage'] = combined_data.obs['age'].apply(age_to_group)
# Optionally convert to categorical with order:
combined_data.obs['development_stage'] = pd.Categorical(
combined_data.obs['development_stage'],
categories=['Prenatal', 'Childhood', 'Young Adult', 'Adult', 'Old', 'Unknown'],
ordered=True
)
combined_data.obs['development_stage'].value_counts()
development_stage
Adult 85274
Young Adult 34086
Childhood 0
Prenatal 0
Old 0
Unknown 0
Name: count, dtype: int64
ref = pd.read_csv("../../../../Ensembl_symbol_Human_(GRCh38.p14)/mart_export.txt")
ref
| HGNC symbol | Gene stable ID | |
|---|---|---|
| 0 | MT-TF | ENSG00000210049 |
| 1 | MT-RNR1 | ENSG00000211459 |
| 2 | MT-TV | ENSG00000210077 |
| 3 | MT-RNR2 | ENSG00000210082 |
| 4 | MT-TL1 | ENSG00000209082 |
| ... | ... | ... |
| 86363 | SNHG12 | ENSG00000197989 |
| 86364 | TAF12-DT | ENSG00000229388 |
| 86365 | NaN | ENSG00000289291 |
| 86366 | RNU11 | ENSG00000274978 |
| 86367 | NaN | ENSG00000296488 |
86368 rows × 2 columns
# Current gene symbols from var_names
gene_symbols = combined_data.var_names.to_list()
# Create mapping: symbol -> Ensembl ID
symbol_to_ensembl = dict(zip(ref['HGNC symbol'], ref['Gene stable ID']))
# Map gene symbols to Ensembl IDs (or None if not found)
ensembl_ids = [symbol_to_ensembl.get(gene, None) for gene in gene_symbols]
# Create a mask to keep only genes with Ensembl IDs starting with "ENSG"
keep_mask = [ensembl is not None and ensembl.startswith("ENSG") for ensembl in ensembl_ids]
# Filter var and var_names accordingly
combined_data = combined_data[:, keep_mask].copy() # subset cells and genes
# Update var_names to Ensembl IDs for kept genes
combined_data.var['ensembl_id'] = [ensembl_ids[i] for i, keep in enumerate(keep_mask) if keep]
combined_data.var_names = combined_data.var['ensembl_id'].values
combined_data.var['gene_symbol'] = combined_data.var['gene_ids']
combined_data.X.max()
2672.0
if hasattr(combined_data.X, 'sum'):
combined_data.obs['n_counts'] = np.ravel(combined_data.X.sum(axis=1))
else:
combined_data.obs['n_counts'] = combined_data.X.sum(axis=1)
combined_data.obs['cell_type']
AAACCTGAGACAAAGG-1-0 NKT
AAACCTGAGCGTTCCG-1-0 CD8_naive
AAACCTGAGTGTTTGC-1-0 CD4_memory
AAACCTGCAAACTGTC-1-0 NKT
AAACCTGCACAACGTT-1-0 CD4_naive
...
TTTGGTTTCGGAATCT-1-2 Int.mono
TTTGTCAAGGCATGTG-1-2 Int.mono
TTTGTCACAAAGGAAG-1-2 CD14mono
TTTGTCATCAACACTG-1-2 CD14mono
TTTGTCATCACAACGT-1-2 CD14mono
Name: cell_type, Length: 119360, dtype: category
Categories (30, object): ['ASDC', 'B_naive', 'CD4_memory', 'CD4_naive', ..., 'plasma_IgG', 'plasma_IgM', 'plasma_dividing', 'switched memory']
combined_data.write_h5ad('./raw_data_LPS_Private_Haniffa_lab.h5ad') # save the processed data
1.3. Concatenation of public and preprocessed prived data
adata1 = sc.read('./Private_emily/raw_data_LPS_Private_Haniffa_lab.h5ad')
/nfs/users/nfs_a/am74/.cache/pypoetry/virtualenvs/cytomeister-AO4Zveyp-py3.10/lib/python3.10/site-packages/anndata/_core/anndata.py:1756: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
adata2 = sc.read('./Public_Emily/Public_LPS_PP.h5ad')
# Make var_names unique to avoid conflicts during concatenation
adata2.var_names_make_unique()
adata1.var_names_make_unique()
# Concatenate the two datasets
adata = adata1.concatenate(adata2)
/tmp/ipykernel_983311/3094885911.py:1: FutureWarning: Use anndata.concat instead of AnnData.concatenate, AnnData.concatenate is deprecated and will be removed in the future. See the tutorial for concat at: https://anndata.readthedocs.io/en/latest/concatenation.html
adata = adata1.concatenate(adata2)
adata # check the merged data
AnnData object with n_obs × n_vars = 224150 × 13826
obs: 'patient_id', 'time_after_LPS', 'Sex', 'age', 'batch', 'IR_VJ_1_locus_tcr', 'IR_VJ_2_locus_tcr', 'IR_VDJ_1_locus_tcr', 'IR_VDJ_2_locus_tcr', 'IR_VJ_1_cdr3_tcr', 'IR_VJ_2_cdr3_tcr', 'IR_VDJ_1_cdr3_tcr', 'IR_VDJ_2_cdr3_tcr', 'IR_VJ_1_cdr3_nt_tcr', 'IR_VJ_2_cdr3_nt_tcr', 'IR_VDJ_1_cdr3_nt_tcr', 'IR_VDJ_2_cdr3_nt_tcr', 'IR_VJ_1_expr_tcr', 'IR_VJ_2_expr_tcr', 'IR_VDJ_1_expr_tcr', 'IR_VDJ_2_expr_tcr', 'IR_VJ_1_expr_raw_tcr', 'IR_VJ_2_expr_raw_tcr', 'IR_VDJ_1_expr_raw_tcr', 'IR_VDJ_2_expr_raw_tcr', 'IR_VJ_1_v_gene_tcr', 'IR_VJ_2_v_gene_tcr', 'IR_VDJ_1_v_gene_tcr', 'IR_VDJ_2_v_gene_tcr', 'IR_VJ_1_d_gene_tcr', 'IR_VJ_2_d_gene_tcr', 'IR_VDJ_1_d_gene_tcr', 'IR_VDJ_2_d_gene_tcr', 'IR_VJ_1_j_gene_tcr', 'IR_VJ_2_j_gene_tcr', 'IR_VDJ_1_j_gene_tcr', 'IR_VDJ_2_j_gene_tcr', 'IR_VJ_1_c_gene_tcr', 'IR_VJ_2_c_gene_tcr', 'IR_VDJ_1_c_gene_tcr', 'IR_VDJ_2_c_gene_tcr', 'IR_VJ_1_junction_ins_tcr', 'IR_VJ_2_junction_ins_tcr', 'IR_VDJ_1_junction_ins_tcr', 'IR_VDJ_2_junction_ins_tcr', 'has_ir_tcr', 'multi_chain_tcr', 'IR_VJ_1_locus_bcr', 'IR_VJ_2_locus_bcr', 'IR_VDJ_1_locus_bcr', 'IR_VDJ_2_locus_bcr', 'IR_VJ_1_cdr3_bcr', 'IR_VJ_2_cdr3_bcr', 'IR_VDJ_1_cdr3_bcr', 'IR_VDJ_2_cdr3_bcr', 'IR_VJ_1_cdr3_nt_bcr', 'IR_VJ_2_cdr3_nt_bcr', 'IR_VDJ_1_cdr3_nt_bcr', 'IR_VDJ_2_cdr3_nt_bcr', 'IR_VJ_1_expr_bcr', 'IR_VJ_2_expr_bcr', 'IR_VDJ_1_expr_bcr', 'IR_VDJ_2_expr_bcr', 'IR_VJ_1_expr_raw_bcr', 'IR_VJ_2_expr_raw_bcr', 'IR_VDJ_1_expr_raw_bcr', 'IR_VDJ_2_expr_raw_bcr', 'IR_VJ_1_v_gene_bcr', 'IR_VJ_2_v_gene_bcr', 'IR_VDJ_1_v_gene_bcr', 'IR_VDJ_2_v_gene_bcr', 'IR_VJ_1_d_gene_bcr', 'IR_VJ_2_d_gene_bcr', 'IR_VDJ_1_d_gene_bcr', 'IR_VDJ_2_d_gene_bcr', 'IR_VJ_1_j_gene_bcr', 'IR_VJ_2_j_gene_bcr', 'IR_VDJ_1_j_gene_bcr', 'IR_VDJ_2_j_gene_bcr', 'IR_VJ_1_c_gene_bcr', 'IR_VJ_2_c_gene_bcr', 'IR_VDJ_1_c_gene_bcr', 'IR_VDJ_2_c_gene_bcr', 'IR_VJ_1_junction_ins_bcr', 'IR_VJ_2_junction_ins_bcr', 'IR_VDJ_1_junction_ins_bcr', 'IR_VDJ_2_junction_ins_bcr', 'has_ir_bcr', 'multi_chain_bcr', 'scr_th', 'doublet_scores', 'cal_th', 'scr_predicted_doublets', 'cal_dobulets', 'data_batch', 'individual_id', 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'cell_typist_full_predicted_labels', 'cell_typist_full_majority_voting', 'cell_typist_initial_predicted_labels', 'cell_typist_initial_majority_voting', 'final_anno', 'timeline', 'sample_id', 'all_T', 'final_anno1', 'all_B', 'log1p_n_genes_by_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'log1p_total_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'n_counts', 'cell_type', 'donor_id', 'study', 'tissue', 'development_stage', 'full_clustering', 'initial_clustering', 'Resample', 'Collection_Day', 'Age_interval', 'Swab_result', 'Status', 'Smoker', 'Status_on_day_collection', 'Status_on_day_collection_summary', 'Days_from_onset', 'Site', 'Worst_Clinical_Status', 'Outcome', 'life_stage'
var: 'gene_ids-0', 'mt-0', 'n_cells_by_counts-0', 'mean_counts-0', 'pct_dropout_by_counts-0', 'total_counts-0', 'GEX-0-0', 'GEX-2-0', 'ribo-0', 'hb-0', 'log1p_mean_counts-0', 'log1p_total_counts-0', 'n_cells-0', 'ensembl_id-0', 'gene_symbol-0', 'ensembl_id-1', 'gene_symbol-1', 'feature_types-1'
adata.obs['time_after_LPS'].value_counts() # check time points available
time_after_LPS
normal 148742
6h_LPS 43810
10h_LPS 20890
90m_LPS 10708
Name: count, dtype: int64
adata.obs['cell_type'].value_counts() # check cell types available
cell_type
CD14mono 23888
CD4_naive 18268
B_naive 15093
CD4.Naive 13829
NK_16hi 13473
CD8_naive 13249
CD4_memory 10259
CD8_EM 8869
gdT 8273
CD8.Naive 8105
CD4.CM 7505
Int.mono 7229
CD83_CD14_mono 6772
CD8.TE 6501
CD4.IL22 6379
MAIT 6042
CD8.EM 5734
NK_CD56dim 4922
CD14mono_M1 4129
CD14_mono 3774
NKT 3240
CD16_mono 3007
Treg 2457
Platelets 2363
NK_56hi 2190
DC2 2024
non-swtiched memory 1583
DC3 1463
B_switched_memory 1395
pDC 1374
CD16mono 1213
switched memory 1158
CD8_CM 1004
B_immature 966
Platelet 862
NK_CD56bright 856
CD4.Tfh 800
B_non-switched_memory 647
B_exhausted 334
non-switched memory/CD11c 301
RBC 301
HSPC 227
NK_prolif 221
ILC1_3 213
C1_CD16_mono 198
CD4.EM 188
Plasma_cell_IgA 173
Plasma_cell_IgG 168
DC1 164
plasma_IgA 160
CD4.Th1 101
Plasmablast 95
Plasma_cell_IgM 84
HSC_erythroid 74
CD8.Prolif 70
HSC_CD38pos 61
plasma_dividing 50
plasma_IgG 31
ASDC 23
plasma_IgM 18
Name: count, dtype: int64
# Define a mapping dictionary for renaming
cell_type_mapping = {
# Monocytes
"CD14mono": "CD14 monocytes",
"CD14_mono": "CD14 monocytes",
"CD83_CD14_mono": "CD14 monocytes",
"CD14mono_M1": "CD14 monocytes",
"CD16_mono": "CD16 monocytes",
"CD16mono": "CD16 monocytes",
"C1_CD16_mono": "CD16 monocytes",
# CD4 T cells
"CD4_naive": "CD4+ T cells",
"CD4.Naive": "CD4+ T cells",
"CD4_memory": "CD4+ T cells",
"CD4.CM": "CD4+ T cells",
"CD4.Tfh": "CD4+ T cells",
"CD4.Th1": "CD4+ T cells",
"CD4.IL22": "CD4+ T cells",
"CD4.EM": "CD4+ T cells",
# CD8 T cells
"CD8_naive": "CD8+ T cells",
"CD8.Naive": "CD8+ T cells",
"CD8_EM": "CD8+ T cells",
"CD8.EM": "CD8+ T cells",
"CD8_CM": "CD8+ T cells",
"CD8.TE": "CD8+ T cells",
"CD8.Prolif": "CD8+ T cells",
# NK cells
"NK_16hi": "NK",
"NK_56hi": "NK",
"NK_CD56dim": "NK",
"NK_CD56bright": "NK",
"NK_prolif": "NK",
# B cells
"B_naive": "B cell",
"B_switched_memory": "B cell",
"non-swtiched memory" : "B cell",
"switched memory": "B cell",
"B_non-switched_memory": "B cell",
"non-switched memory": "B cell",
"non-switched memory/CD11c": "B cell",
"B_immature": "B cell",
"B_exhausted": "B cell",
# Plasma cells
"Plasma_cell_IgA": "plasma cell",
"plasma_IgA": "plasma cell",
"Plasma_cell_IgG": "plasma cell",
"plasma_IgG": "plasma cell",
"Plasma_cell_IgM": "plasma cell",
"plasma_IgM": "plasma cell",
"Plasmablast": "plasma cell",
"plasma_dividing": "plasma cell",
# Others
"Platelets": "platelet",
"Platelet": "platelet",
"Treg": "regulatory T cell",
"MAIT": "mucosal invariant T cell",
"gdT": "gamma-delta T cell",
"NKT": "NKT",
"pDC": "Plasmocytoid dendritic cell",
"DC1": "Dendritic cells",
"DC2": "Dendritic cells",
"DC3": "Dendritic cells",
"ASDC": "ASDC",
"Int.mono": "CD14 monocytes",
"RBC": "Red_Blood_Cell",
"HSPC": "hematopoietic stem cell",
"HSC_erythroid": "HSC_Erythroid",
"HSC_CD38pos": "HSC_CD38pos",
"ILC1_3": "ILC1_3",
}
# Apply the mapping to adata.obs['cell_type']
adata.obs['cell_type_harmonized'] = adata.obs['cell_type'].map(cell_type_mapping).fillna(adata.obs['cell_type'])
# Verify the new counts
print(adata.obs['cell_type_harmonized'].value_counts())
cell_type_harmonized
CD4+ T cells 57329
CD14 monocytes 45792
CD8+ T cells 43532
NK 21662
B cell 21477
gamma-delta T cell 8273
mucosal invariant T cell 6042
CD16 monocytes 4418
Dendritic cells 3651
NKT 3240
platelet 3225
regulatory T cell 2457
Plasmocytoid dendritic cell 1374
plasma cell 779
Red_Blood_Cell 301
hematopoietic stem cell 227
ILC1_3 213
HSC_Erythroid 74
HSC_CD38pos 61
ASDC 23
Name: count, dtype: int64
# Get unique timepoints
all_timepoints = adata.obs['time_after_LPS'].unique()
# Group by cell_type and get unique timepoints per cell type
cell_type_timepoints = (
adata.obs
.groupby('cell_type_harmonized')['time_after_LPS']
.unique()
.apply(list)
)
# Find cell types that do not appear in all timepoints
missing_in_any_timepoint = [
ct for ct in cell_type_timepoints.index
if not set(all_timepoints).issubset(set(cell_type_timepoints[ct]))
]
print("Cell types not present in all timepoints:", missing_in_any_timepoint)
Cell types not present in all timepoints: ['ASDC', 'HSC_CD38pos', 'HSC_Erythroid', 'ILC1_3', 'Red_Blood_Cell']
adata = sc.read('./full_lps.h5ad')
cell_types_to_remove = ['ASDC', 'HSC_CD38pos', 'HSC_Erythroid', 'ILC1_3', 'Red_Blood_Cell']
keep_mask = ~adata.obs['cell_type_harmonized'].isin(cell_types_to_remove)
adata_filtered = adata[keep_mask, :].copy()
adata_filtered.obs['cell_type_harmonized'].value_counts()
cell_type_harmonized
CD4+ T cells 57329
CD14 monocytes 45792
CD8+ T cells 43532
NK 21662
B cell 21477
gamma-delta T cell 8273
mucosal invariant T cell 6042
CD16 monocytes 4418
Dendritic cells 3651
NKT 3240
platelet 3225
regulatory T cell 2457
Plasmocytoid dendritic cell 1374
plasma cell 779
hematopoietic stem cell 227
Name: count, dtype: int64
len(set(adata_filtered.obs['cell_type_harmonized']))
15
adata_filtered.X.max()
np.float32(4066.0)
adata_filtered.write_h5ad('./full_lps.h5ad') # save the final processed data